Part 2: Cardiac Electrophysiology
Wed Jul 08 2026
By B. Hassan

Electrophysiology
The sarcolemma of each of these cardiac cell types is a phospholipid bilayer that is largely impermeable to ions. There are transport proteins interspersed throughout the membrane that help maintain ionic concentration gradients between the inside and the outside of the cardiac cells.
Normally the Na+ and Ca2+ concentrations are much higher outside the cell, and the K+ concentration is much higher inside. This is attributed to the cell membrane transporter Na+/K+-ATPase, which actively (i.e. ATP dependent) pumps 3 Na+ ions out of the cell in exchange for the inward movement of 2 K+ ions.
The movement of ions across the cell membrane serves as the basis of the action potential. Passive ion movement depends on two major actors:
- The energetic favorability
- The permeability of the membrane for the ion
Energetic favorability
Molecules diffuse from areas of high concentration to areas of lower concentration, and the gradient between these values determines the flow rate of ions. For example, the extracellular Na+ concentration is normally 145 mM, while the intracellular concentration is 15 mM. As a result, a strong diffusive force tends to drive Na+ into the cell, down its concentration gradient.
The transmembrane potential of a myocyte at rest is about −90 mV, meaning compared to the extracellular space, the inside of a myocyte is negative. Extracellular Na+ (positively charged) is therefore attracted to the relatively negatively interior of the cell.
Thus, there is a strong tendency or Na+ to enter the cell and remain there, because of the steep concentration gradient and the electrical attraction.
Permeability
The membrane of the cell at its resting potential is not permeable to sodium. The hydrophobic phospholipid bilayer of the cell membrane does not allow simple passage of charged, hydrophilic particles. This means that even if there is a strong force driving Na+ into the cell, the ions can’t normally get inside.
Instead, permeability of the membrane is dependent on the opening of ion channels, which are specialized transport proteins that span the cell membrane, allowing for certain charged molecules to pass under specific conditions. Each type of channel is normally selective for a specific ion (e.g. sodium or potassium channels) depending on the size and structure of the channel.
Ion channels are gated, meaning that at any given moment, a channel can be either open or closed, and ions can only pass when the channel is open. The more time a channel is open for, the larger the number of ions that can pass through it.
The voltage across the membrane (i.e. the membrane potential) determines what fraction of channels is open at a given time. Therefore, the gating of channels is said to be voltage sensitive. As the membrane voltage changes during depolarization and repolarization of the cell, specific channels open and close.
Example
An example of voltage-sensitive gating is the fast sodium channels of the heart. The transmembrane protein that forms this channel assumes different conformations depending on the cell’s membrane potential. At a voltage of −90 mV (the typical resting membrane potential of a cardiac muscle cell), the channels are predominantly closed such that Na+ ions cannot pass through.
A rapid wave of depolarization renders the membrane potential less negative. This activates the closed fast sodium channels to change conformation to the open state, allowing Na+ ions to move inside the cell, producing an inward current that depolarizes the cell, reducing the negativity of the cell membrane potential. However, the activated channels remain open only for a brief time and then spontaneously close to an inactive state. The inactivated state persists until the membrane voltage has been repolarized nearly back to its original resting level, after which, the channels transition to the closed but ready-to-open state.
Another important attribute of cardiac fast sodium channels is that if the membrane potential of a cardiac cell is slowly depolarized and maintained chronically at levels less negative than the usual resting membrane potential potential, inactivation of channels occurs without an initial opening. As long as this partial depolarization exists, the closed, inactive channels cannot recover to the resting state. This is the typical case in cardiac pacemaker cells, in which the membrane voltage is generally less negative than −70 mV throughout the cardiac cycle. As a result, the fast sodium channels in pacemaker cells are persistently inactivated and do not play a role in the generation of the action potential.
Note
Technically speaking, most pacemaker cells lack the fundamental expression of fast sodium channels, and for the tiny amount that do have them, they are kept in a permanently inactive state.
Resting potential
This is the electrical potential difference between the inside and outside of a cell in a resting state prior to any excitation.
Cardiac myocytes contain a set of inward rectifier K+ channels that are open in the resting state, at a time when other ion channels are closed. This makes the resting cell membrane much more permeable to potassium than to other ions. As a result, K+ flows in an outward direction down its concentration gradient, removing positive charges from the cell. As K+ exit the cell, negatively charged anions that cannot exit the cell (impermeable to passage) remain inside, and so the interior of the cell becomes negatively charged relative to the outside.
As the interior of the cell becomes more negatively charged by the outward flow of potassium, the positively charged K+ ions are attracted back toward the negatively charged cell interior, slowing their exit from the cell (i.e. the K+ concentration gradient and the electrostatic force oppose each other). At equilibrium, these forces cancel each other out, resulting in zero net movement of K+ across the cell membrane. The electrical potential at which that equilibrium is reached is known as the potassium equilibrium potential and is −91 mV in myocytes. Since at rest the membrane is almost exclusively permeable to potassium, this value closely approximates the cell’s resting potential.
The potassium equilibrium potential is calculated using the Nernst equation:
Potassium equilibrium potential
The permeability of the cardiac cell membrane for sodium is minimal in the resting state because sodium channels are essentially closed. However, there is a slight leak of sodium ions into the cell at rest. This small inward current of positively charged ions explains why the actual resting potential is slightly less negative (−90 mV) than would be predicted by the Nernst equation.
The sodium ions that slowly leak into the myocyte at rest and the much larger amount that enters during the action potential are continuously removed from the cell and returned to the extracellular environment by the Na+/K+-ATPase.
Cardiomyocytes action potential
There are 3 broad categories of cardiac myocytes depending on their electrophysiologic properties:
- Pacemaker cells (e.g., SA node, AV node)
- Specialized rapidly conducting fibers (i.e. Purkinje network)
- Contractile muscle cells
Each of these three cell types expresses a unique configuration of ion channels, creating 3 specialized action potentials tailored perfectly to their specific functions.
Cardiac Muscle Cell
The action potential of cardiac muscle cells does not normally occur spontaneously. Instead, when a wave of depolarization reaches the myocyte through the gap junctions with neighboring cells, its membrane potential becomes less negative, triggering an action potential. These gap junctions are a special type of direct intercellular communication channels that provide electrical and biochemical coupling between cells.
The resting membrane potential of cardiac myocytes remains stable at -90 mV. This resting state before any depolarization is known as phase 4 of the cardiac myocyte action potential.
Other than phase 4, there are 3 other phases that characterize the cardiac myocyte action potential:
Phase 0
At phase 4, sodium and calcium channels are closed. An initial wave of depolarization decreases the negativity of the resting membrane potential, causing opening of some of the fast sodium channels, causing sodium in the extracellular space to rush inside the cell down its concentration and electrochemical gradients (since the positive sodium ions are attracted to the relatively negative intracellular space).
The entry of sodium ions inside the cell causes the membrane potential to become progressively less negative, causing more sodium channels to open, and in turn more sodium ions to enter the cell. When the membrane potential reaches a special threshold of approximately −70 mV, enough of the fast sodium channels have opened to generate a self-sustaining inward Na+ current (termed ). The entry of sodium ions at and beyond that threshold exceeds the exit of K+ ions through the open inward rectifier channels, such that a positive feedback loop starts, and the cell continues to depolarize.
This influx of sodium ions can be seen on a time-membrane potential graph as a rapid upstroke known as phase 0 or rapid depolarization, taking the cell membrane potential in the positive range, and reaching a peak of approximately +20 mV to +50 mV. However, the sodium channels remain open only for a brief moment before quickly transitioning to an inactivated state, preventing further influx of sodium and further depolarization.
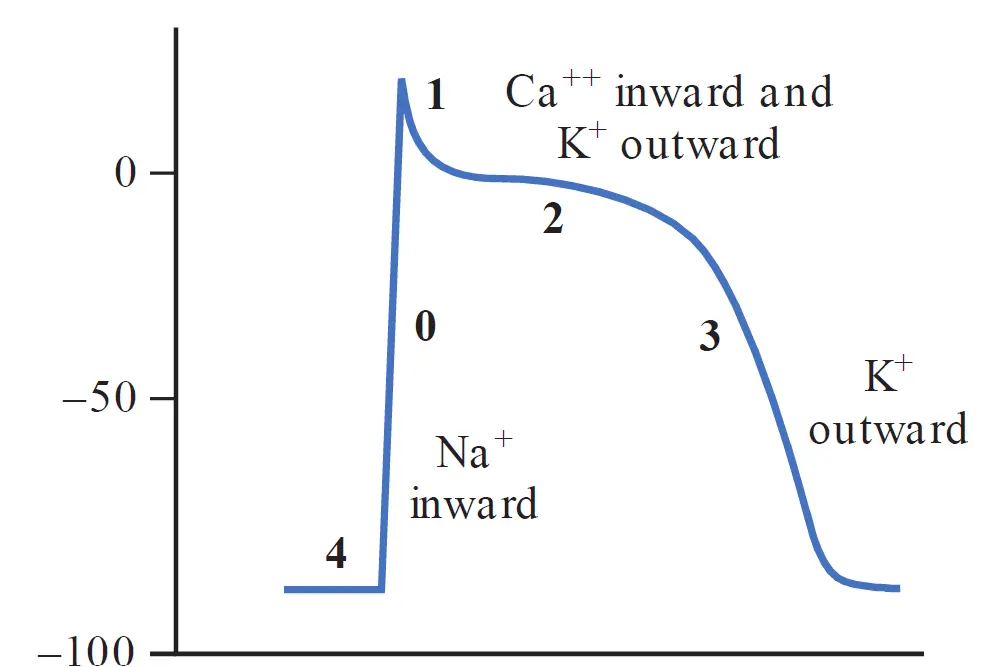
Phase 1
Following rapid phase 0, a brief current of repolarization (termed ) returns the membrane potential to approximately 0 mV. This happens by the opening of a transiently activated potassium channel, causing a brief outflow of K+ ions.
Phase 2
This relatively long plateau phase is mediated by a balance between the outward potassium outflow (known as and ) carried through the voltage-gated delayed rectifier K+ channels and an inward Ca2+ (termed ) inflow carried by L-type calcium channels. This balance results in nearly zero net current, and the membrane potential does not change for a prolonged period, which accounts for the flat plateau portion of the action potential curve.
Note
The L in L-type calcium channels stands for long-lasting, because these channels open for a long period of time, which characterizes the long duration of the plateau phase.
The L-type calcium channels begin to open during phase 0 when the membrane potential reaches approximately −40 mV, however, this activation than the activation of the fast sodium channels, and so the influx of Ca2+ proceeds in a more gradual manner compared to the influx of Na+ ions.
Phase 3
As L-type Ca2+ channels gradually inactivate and the outflow of K+ begins to exceed the inflow of calcium, phase 3 begins, and rapid repolarization returns the membrane potential back to the resting membrane potential (−90 mV), preparing the cell or the next stimulus or depolarization.
To preserve normal transmembrane ionic concentration gradients, sodium and calcium ions that enter the cell during depolarization must be returned to the extracellular environment, and potassium ions must return to the cell interior.
- Ca2+ ions are removed by the sarcolemmal Na+/Ca2+ exchanger, and to a lesser extent by the ATP-energized calcium pump (sarcolemmal Ca2+ ATPase).
- The corrective exchange of Na+ and K+ across the cell membrane is mediated by Na+/K+-ATPase, as described earlier.
Pacemaker Cells
Those cells exhibit an important property called automaticity, meaning they spontaneously depolarize without the need of an external stimulator, and they set the heart rate such that the action potentials of all other cardiac cells follow the action potentials of the pacemaker cells.
Cells that normally display pacemaker behavior include the SA node (the “natural pacemaker” of the heart) and the AV node. Although muscle cells do not normally display automaticity, they may do so under disease conditions such as ischemia.
The shape of the action potential of a pacemaker cell is different from that of a ventricular muscle cell in:
-
The maximum negative voltage of pacemaker cells is approximately −60 mV, substantially less negative than the resting potential of muscle cells (−90 mV). The persistently less negative membrane voltage of pacemaker cells causes persistent inactivation of fast sodium channels in these cells.
-
Phase 4 of the pacemaker cell action potential is not flat, but has an upward slope representing spontaneous gradual depolarization. This spontaneous depolarization is the result of sodium influx from a special types of sodium channels called funny channels, or pacemaker channels, producing a Na+ influx known as the pacemaker current (denoted by ). These funny channels are different from the fast sodium channels responsible for phase 0 of the action potential. Importantly, these funny channels opens in the very negative voltage ranges reached during repolarization of the cell. The influx of Na+ through the pacemaker channel causes the membrane potential to become progressively less negative during phase 4, ultimately depolarizing the cell to its threshold voltage.
-
The phase 0 upstroke of the pacemaker cell action potential is less rapid and reaches a lower amplitude than that of a cardiac muscle cell. This results from the absence of fast sodium channels and the upstroke of the action potential completely relying on Ca2+ influx through the relatively slow L-type calcium channels.
Note
Funny channels are also called hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, which describes their most important property, which is that they open in the very negative voltage ranges reached during repolarization of the cell
Repolarization of pacemaker cells occurs in similar fashion to that of cardiac muscle cells; relying on the inactivation of the L-type calcium channels and increased activation of potassium channels.
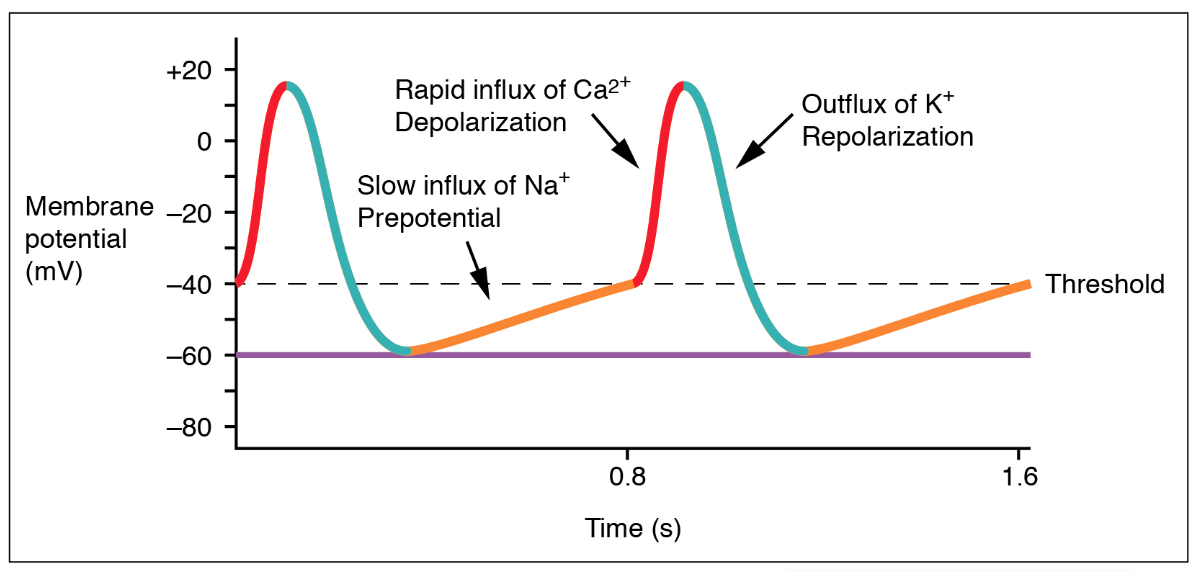
Heart rate
Normally, the heart follows the source of the highest rhythm as the rapid electrical waves spread from that source of rhythm, constantly resetting other sources of rhythm to its own rate. This is called overdrive suppression.
In a healthy heart, the SA node fires the fastest at approximately 70 bpm and forces its rhythm upon the rest of the heart. The AV node (~50 bpm) and the Purkinje network (~30 bpm) exhibit automaticity and can serve as their own pacemakers. However, the rapid electrical waves rushing down from the SA node constantly resets them before they can reach their own threshold.
Certain diseases affect the SA node, preventing this constant inhibition, and allowing the AV node to take over as the pacemaker of the heart, causing the whole heart to follow the AV nodal rhythm.
Refractory period
Compared with action potential in nerves and skeletal muscle, the cardiac action potential is much longer in duration, supporting prolonged Ca2+ entry and muscle contraction during systole. This results in a prolonged period of channel inactivation during which the muscle is refractory (unresponsive) to restimulation. Such a long refractory period is important because it allows the ventricles sufficient time to relax and refill before the next contraction.
There are different levels of refractoriness during the action potential of a myocyte. The degree of refractoriness primarily reflects the percentage of fast sodium channels that have recovered from the inactive state and are capable of reopening. As phase 3 of the cardiac muscle cell action potential progresses, an increasing number of fast sodium channels recover from the inactivated to resting states, and can then open in response to the next depolarization wave. This, in turn, corresponds to an increased probability that a stimulus will trigger an action potential and result in a propagated impulse.
During the absolute refractory period (ARP), the cell is completely unresponsive to stimulation. The ARP includes most of phase 0, all of phase 1, and all of phase 2.
The effective refractory period includes a brief time beyond the ARP (extends to include a short interval of phase 3) during which stimulation produces a localized depolarization that does not propagate.
The relative refractory period is the interval during which stimulation triggers an action potential that is conducted, but the slope of phase 0 is lower during that second action potential as some of the fast sodium channels remain in the inactivated state, reducing the available net inward current.
Following the relative refractory period, a short “supranormal” period is present, occurring at the very end of Phase 3. At this phase, a less-than-normal stimulus can trigger an action potential. This occurs as enough fast sodium channels have recovered to initiate an action potential. At the same time, the cell membrane potential is less negative that at the resting phase, meaning a smaller than normal stimulus can push the cell to the -70 mV threshold required to initiate an action potential.
Note
Working ventricular muscle cells do not display a true supranormal phase under normal conditions. This is because Purkinje fibers have a prolonged, gradual exit out of Phase 3 because of the specific slow kinetics of their outward potassium currents. This slow “landing” stretches out that “supranormal” period interval. Muscle cells, on the other hand, slam back down to their resting potential so quickly that the threshold and recovery lines cross without creating a functional supranormal window. However, in certain heart diseases where conduction is depressed, an electrical impulse arriving at just the right time during this supranormal phase can unexpectedly conduct, which may initiate a dangerous arrhythmia.
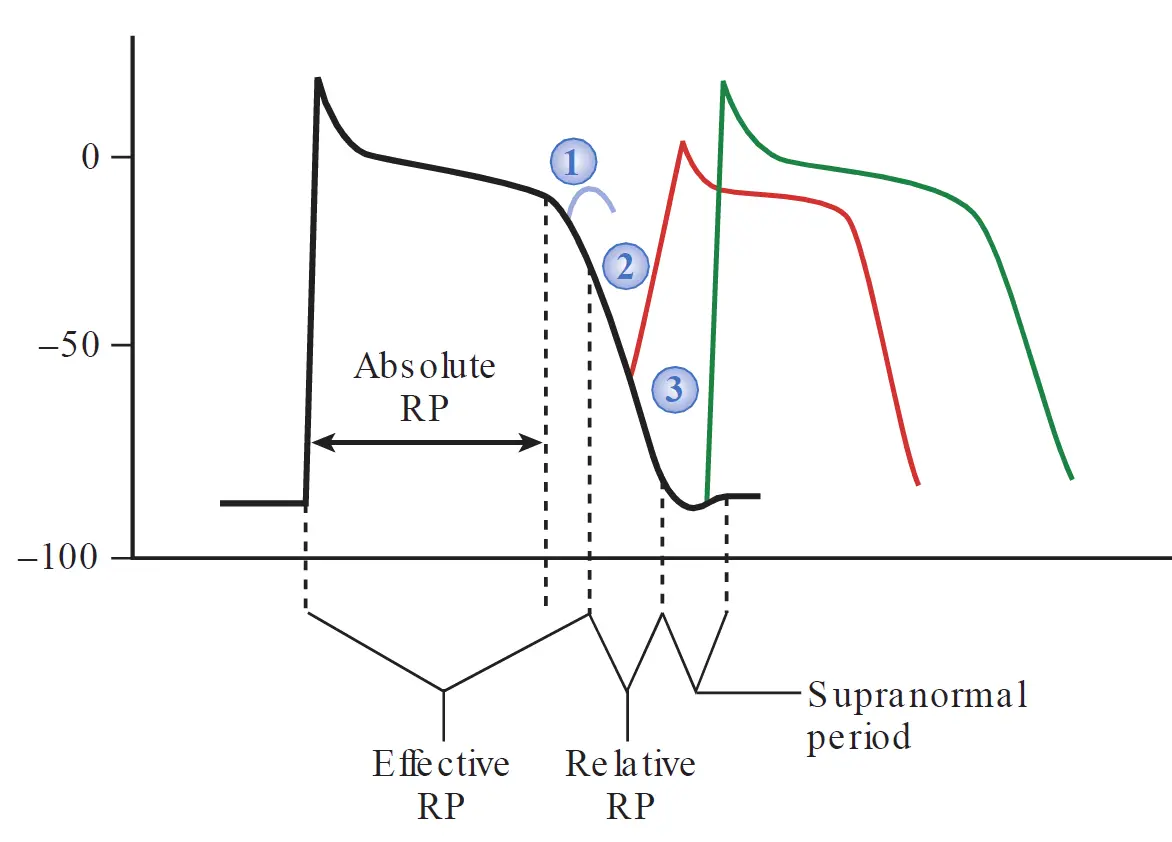
The refractory period of atrial cells is shorter than that of ventricular muscle cells, such that atrial rates can generally exceed ventricular rates during rapid arrhythmias.
Factors affecting conductivity
In cardiac myocytes, the depolarization wave spreads rapidly from cell-to-cell as each myocyte is connected to its neighbors through gap junctions. The speed of conduction of a cardiac myocyte depends on:
-
The number of sodium channels in the myocyte membrane.
-
The value of the resting membrane potential. The less negative the resting membrane potential is, the greater the fraction of fast sodium channels that are in the inactivated state and the less rapid the upstroke velocity.
-
The amount of gap junctions between the myocyte and its neighbors.
Tissue specialized for rapid conduction like the Purkinje fibers have a high concentration of fast sodium channels and a is highly connected with its neighbors through gap junctions, allowing its action potential to spread quickly between cells.
Sequence of cardiac depolarization
Electrical activation of a heartbeat normally begins at the SA node, which exhibits automaticity as we discussed earlier. The impulse then spreads to the surrounding atrial muscles through the gap junctions. Atrial muscles participate in the propagation of the action potential from the SA to the AV node.
The highly fibrous cardiac skeleton that surrounds the mitral and tricuspid valves electrically disconnects the atria from the ventricles, making it such that there is no direct electrical connection between the atria and the ventricles other than the AV node.
As the action potential reaches the AV node, a delay in conduction of approximately 0.1 seconds occur. This is because fibers of the AV node have a small dimeter, have few gap junctions with its neighbors, and have no fast sodium channels (recall that fast sodium channels are either non-existent or permanently inactivated in pacemaker tissue).
This delayed conduction at the AV node gives the atria to fully empty their blood into the ventricles before ventricular stimulation. Moreover, it is also critical for protecting the ventricles from rapid atrial arrhythmias.
Note
The AV node has a maximum conduction capacity of approximately 180 to 230 impulses per minute. During rapid atrial arrhythmias, this limit acts as a protective electrical filter, preventing excessively high atrial rates from conducting to the ventricles and shielding the heart from ventricular arrhythmias or arrest.
After traversing the AV node, the action potential spreads into the rapidly conducting bundle of His and Purkinje fibers, which transmit the electrical impulses to the bulk of the ventricular muscles in a spatially synchronized manner. This allows for organized contraction of ventricular myocytes as one unit, optimizing the volume of blood pumped by the heart.
Excitation-contraction coupling
Excitation–contraction coupling is the process by which an action potential translates to a physical contraction of cardiac muscles.
Earlier, we have described the histology of the heart muscle cells, which is important to understand the following section.
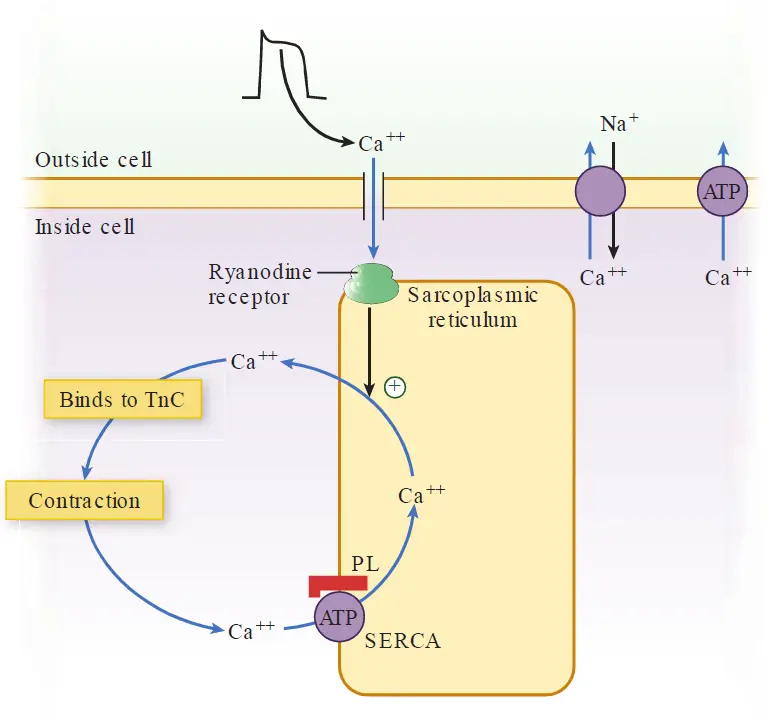
Calcium-induced calcium release
The cycling of calcium ions in and out of the cell cytosol during each action potential effectively couples an electrical impulse to physical muscle contraction.
Recall that during phase 2 of the action potential, activation of L-type Ca++ channels results in an influx of Ca2+ ions into the myocyte. The small amount of calcium that enters the cell through these channels is not sufficient to initiate myocyte contraction. However, this small amount of calcium influx triggers a much greater release of Ca2+ from the terminal cisterns of myocyte SR, which serve as abundant stores of calcium. This occurs through specialized Ca2+ receptors in the SR called ryanodine receptors.
This amplification of the initial Ca2+ influx through the L-type calcium channels is called calcium induced calcium release (CICR).
As Ca2+ bind to TnC, the activity of TnI is inhibited, which induces a conormational change in tropomyosin, freeing the actin active sites and allowing the sliding of actin over myosin in an ATP dependent manner, initiating the contraction process.
The first step in this process is activation of the myosin head by hydrolysis of ATP, forcing myosin to change shape into a high-energy position (like pulling back the string on a crossbow). Now that the myosin head is energized and the active sites of actin are free, actin can bind to myosin. This binding induces a conformational change in the myosin heads, causing the heads to snap back to its relaxed angle, dragging the actin filament along with it, which is known as a power stroke.
Now, the myosin head is stuck against the actin filament at the end of its stroke. To detach so it can repeat the process, a new ATP molecule must bind to the myosin head. Once detached, the myosin head hydrolyzes that new ATP, energizes itself again, and the cycle repeats.
This cycle can be visualized as a sliding process, in which binding of actin to myosin generates a power stroke, resulting in a small movement of the actin over the myosin. As the cycle repeats, further power strokes allow further movement of the actin along the myosin increases the overlap between the myofilaments, and shortening of overall Z to Z distance, which is seen as a muscle contraction. In the presence of ATP, this process continues for as long as the cytosolic calcium concentration remains sufficiently high to inhibit the troponin–tropomyosin blocking action.
Myocyte relaxation, like contraction, is synchronized with the electrical activity of the cell. At the end of phase 2 of the action potential, L-type channels inactivate, stopping the influx of Ca2+ and abolishing the trigger for calcium-induced calcium release.
Concurrently, calcium is pumped back into the SR by sarco(endo)plasmic reticulum Ca++ ATPase or SERCA. Moreover, the small amount of Ca2+ that entered the cell through L-type calcium channels is removed via the sarcolemmal Na+/Ca2+ exchanger and to a lesser extent by the sarcolemmal Ca++-ATPase.
As cytosolic Ca2+ concentrations fall and calcium ions dissociate from TnC, tropomyosin once again inhibits the actin–myosin interaction, leading to relaxation of the cell. The contraction–relaxation cycle can then repeat within the next action potential.
β-Adrenergic and Cholinergic Signaling
There is substantial evidence that the concentration of Ca2+ within the cytosol is the major determinant of the force of cardiac contraction with each heartbeat. Mechanisms that raise intracellular Ca2+ concentration enhance contractile force, and vice versa.
β-Adrenergic stimulation is one mechanism that increases Ca2+ concentration within myocytes, thus increasing the force of contraction. Catecholamines, mainly norepinephrine bind to the cardiac β1-adrenergic receptors, which is a G protein receptor (Gs) that stimulates membrane-bound adenylate cyclase that converts ATP to cyclic AMP (cAMP). cAMP in turn activates specific protein kinases that phosphorylate cellular proteins, including L-type calcium channels. This increases the channel Ca2+ influx, increasing calcium induced calcium release which in turn increases the force of contraction.
β-Adrenergic stimulation also enhances myocyte relaxation. The return of Ca2+ from the cytosol to the SR is regulated by phospholamban (PL), a protein in the SR membrane. In its dephosphorylated state, PL inhibits Ca2+ uptake by SERCA. However, β-adrenergic activation of protein kinase causes PL to become phosphorylated, reducing its inhibitory effect. This increases Ca2+ uptake by SR, increasing clearance of Ca2+ from cytosol and promoting myocyte relaxation. The increased cAMP activity also results in phosphorylation of TnI, inhibiting actin–myosin interactions and further enhancing myocyte relaxation.
Cholinergic signaling via parasympathetic inputs opposes the effects of β-adrenergic stimulation.
Acetylcholine released from parasympathetic nerves (mainly from the vagus nerve) binds to the muscarinic M2 receptor on cardiac cells. This receptor also activates a coupled protein (Gi) that opposes the action of Gs protein and inhibits adenylate cyclase. At the SA node, the action of cholinergic stimulation serves to reduce heart rate, by reducing the slope of phase 4, reducing the automaticity of pacemaker cells. In the myocardium, the effect is to counteract the force of contraction induced by β-adrenergic stimulation.
Ventricular escape
Note that ventricular cells are much less sensitive to cholinergic effects than atrial cells, in a process called vagal escape, which likely owes to the fact that anatomically, the vagus nerve heavily coats the SA and AV nodes and the atrial muscle, while the ventricular conduction system (the His-Purkinje network) has almost no vagal innervation. This serves to protect the ventricle from abnormally high vagal stimuli.
For example, when the vagus nerve produces an abnormally large cholinergic discharge, both the SA and the AV nodes may undergo arrest and stop generating electrical impulses. If this happens, the funny channels within the Purkinje fibers may slowly and naturally leak sodium inside the cells, causing the membrane potential to slowly drift upwards until it hits its own threshold, creating a spontaneous action potential.
Note
Gs means stimulatory G protein, while Gi means inhibitory G protein